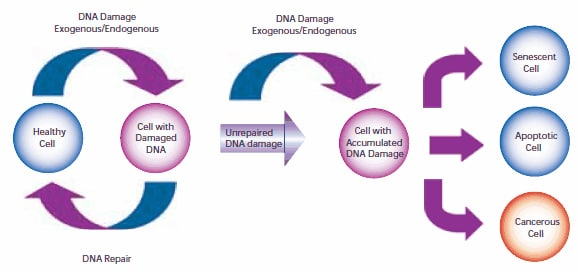
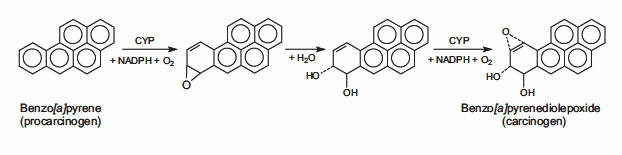
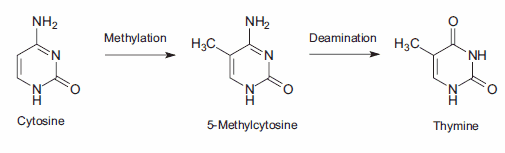
14.Müller A, Fishel R. 2002. Mismatch Repair and the Hereditary Non-polyposis Colorectal Cancer Syndrome (HNPCC). Cancer Investigation. 20(1):102-109. https://doi.org/10.1081/cnv-120000371
15.Lindahl T. 1974. An N-Glycosidase from Escherichia coli That Releases Free Uracil from DNA Containing Deaminated Cytosine Residues. Proceedings of the National Academy of Sciences. 71(9):3649-3653. https://doi.org/10.1073/pnas.71.9.3649
16.Singer B, Hang B. 1997. What Structural Features Determine Repair Enzyme Specificity and Mechanism in Chemically Modified DNA??. Chem. Res. Toxicol.. 10(7):713-732. https://doi.org/10.1021/tx970011e
17.Lindahl T. 1999. Quality Control by DNA Repair. 286(5446):1897-1905. https://doi.org/10.1126/science.286.5446.1897
18.Caldecott KW, Aoufouchi S, Johnson P, Shall S. 1996. XRCC1 Polypeptide Interacts with DNA Polymerase and Possibly Poly (ADP-Ribose) Polymerase, and DNA Ligase III Is a Novel Molecular 'Nick-Sensor' In Vitro. Nucleic Acids Research. 24(22):4387-4394. https://doi.org/10.1093/nar/24.22.4387
19.Dantzer F, de la Rubia G, Ménissier-de Murcia J, Hostomsky Z, de Murcia G, Schreiber V. 2000. Base Excision Repair Is Impaired in Mammalian Cells Lacking Poly(ADP-ribose) Polymerase-1?. Biochemistry. 39(25):7559-7569. https://doi.org/10.1021/bi0003442
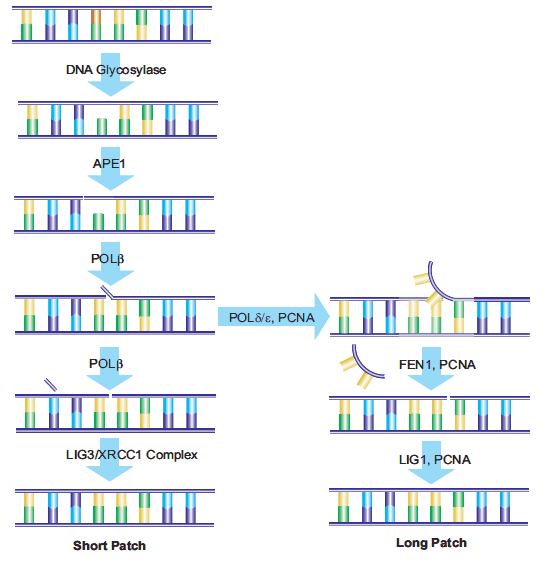
20.Srivastava DK, Vande Berg BJ, Prasad R, Molina JT, Beard WA, Tomkinson AE, Wilson SH. 1998. Mammalian Abasic Site Base Excision Repair. J. Biol. Chem.. 273(33):21203-21209. https://doi.org/10.1074/jbc.273.33.21203
21.Ranalli TA, Tom S, Bambara RA. 2002. AP Endonuclease 1 Coordinates Flap Endonuclease 1 and DNA Ligase I Activity in Long Patch Base Excision Repair. J. Biol. Chem.. 277(44):41715-41724. https://doi.org/10.1074/jbc.m207207200
22.Sattler U, Frit P, Salles B, Calsou P. 2003. Long?patch DNA repair synthesis during base excision repair in mammalian cells. EMBO Rep. 4(4):363-367. https://doi.org/10.1038/sj.embor.embor796
23.Fortini P, Pascucci B, Parlanti E, Sobol RW, Wilson SH, Dogliotti E. 1998. Different DNA Polymerases Are Involved in the Short- and Long-Patch Base Excision Repair in Mammalian Cells?. Biochemistry. 37(11):3575-3580. https://doi.org/10.1021/bi972999h
24.Klungland A. 1997. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). 16(11):3341-3348. https://doi.org/10.1093/emboj/16.11.3341
25.Sung J, Demple B. 2006. Roles of base excision repair subpathways in correcting oxidized abasic sites in DNA. FEBS Journal. 273(8):1620-1629. https://doi.org/10.1111/j.1742-4658.2006.05192.x
26.Balajee AS, Bohr VA. 2000. Genomic heterogeneity of nucleotide excision repair. Gene. 250(1-2):15-30. https://doi.org/10.1016/s0378-1119(00)00172-4
27.Gros L, Saparbaev MK, Laval J. 2002. Enzymology of the repair of free radicals-induced DNA damage. Oncogene. 21(58):8905-8925. https://doi.org/10.1038/sj.onc.1206005
28.You J, Wang M, Lee S. 2003. Biochemical Analysis of the Damage Recognition Process in Nucleotide Excision Repair. J. Biol. Chem.. 278(9):7476-7485. https://doi.org/10.1074/jbc.m210603200
29.Rosell R, Taron M, Barnadas A, Scagliotti G, Sarries C, Roig B. 2003. Nucleotide Excision Repair Pathways Involved in Cisplatin Resistance in Non-Small-Cell Lung Cancer. Cancer Control. 10(4):297-305. https://doi.org/10.1177/107327480301000404
30.Vogel U, Dybdahl M, Frentz G, Nexø BA. 2000. DNA repair capacity: inconsistency between effect of over-expression of five NER genes and the correlation to mRNA levels in primary lymphocytes. Mutation Research/DNA Repair. 461(3):197-210. https://doi.org/10.1016/s0921-8777(00)00051-3
31.Hanawalt PC. 2002. Subpathways of nucleotide excision repair and their regulation. Oncogene. 21(58):8949-8956. https://doi.org/10.1038/sj.onc.1206096
32.Critchlow SE, Jackson SP. 1998. DNA end-joining: from yeast to man. Trends in Biochemical Sciences. 23(10):394-398. https://doi.org/10.1016/s0968-0004(98)01284-5
33.Wang H. 2003. Biochemical evidence for Ku-independent backup pathways of NHEJ. Nucleic Acids Research. 31(18):5377-5388. https://doi.org/10.1093/nar/gkg728
34.Perrault R, Wang H, Wang M, Rosidi B, Iliakis G. 2004. Backup pathways of NHEJ are suppressed by DNA-PK. J. Cell. Biochem.. 92(4):781-794. https://doi.org/10.1002/jcb.20104
35.Sánchez-Pérez I. 2006. DNA repair inhibitors in cancer treatment. Clin Transl Oncol. 8(9):642-646. https://doi.org/10.1007/s12094-006-0034-8
36.Madhusudan S, Hickson ID. 2005. DNA repair inhibition: a selective tumour targeting strategy. Trends in Molecular Medicine. 11(11):503-511. https://doi.org/10.1016/j.molmed.2005.09.004
37.PLUMMER E. 2006. Inhibition of poly(ADP-ribose) polymerase in cancer. Current Opinion in Pharmacology. 6(4):364-368. https://doi.org/10.1016/j.coph.2006.02.004
38.SABHARWAL A, MIDDLETON M. 2006. Exploiting the role of O6-methylguanine-DNA-methyltransferase (MGMT) in cancer therapy. Current Opinion in Pharmacology. 6(4):355-363. https://doi.org/10.1016/j.coph.2006.03.011